1. There are multiple types of ALS.
They are based on the body parts the disease affects when it initially shows up. Bulbar and Spinal are the two most common forms. Bulbar onset initially affects the muscles controlling speech and swallowing. Spinal onset initially affects muscles controlling the arms and legs. There are several additional less common subtypes of ALS.
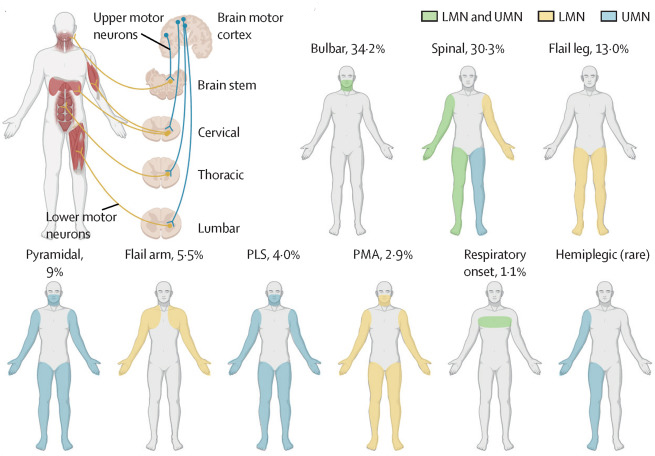
Figures 1 A & B from Dr. Feldman’s ALS Seminar in The Lancet
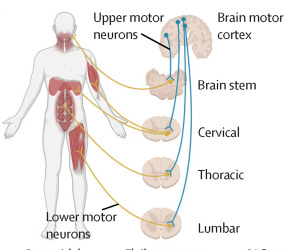
The Lancet, Figure 1 A: a schematic showing UMNs (blue), which relay signals from the motor cortex to the LMNs (yellow), which relay signals to the muscles.
2. At the cellular level, ALS is characterized by degeneration of upper and/or lower motor neurons (nerve cells control muscle contraction).
Upper motor neurons connect the brain to the spinal cord, whereas lower motor neurons connect the spinal cord to the muscles.
3. ALS is progressive and fatal 100% of the time.
Muscle weakness spreads from the initial onset to neighboring muscles, and so forth. This continues until patients are unable to swallow and eventually breathe. The progression of ALS is very individual to each patient.
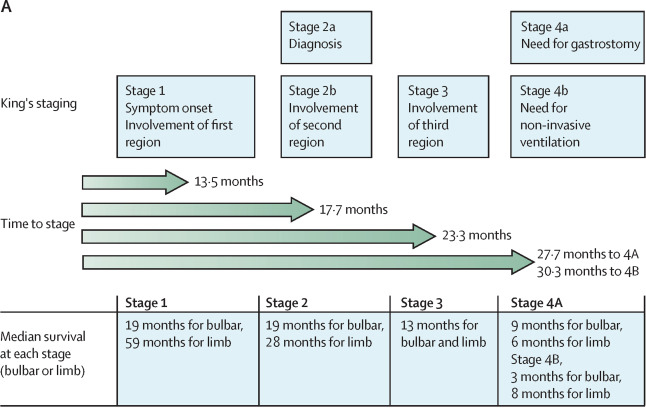
The Lancet, Figure 4 A: King’s staging of ALS, which rates the progression of the disease into four stages
4. In addition to muscle weakness, up to 50% of patients with ALS can also present with changes in cognition and behavior.
These cognitive and behavioral changes support the concept that amyotrophic lateral sclerosis is a global neurodegenerative disease along the same continuum as frontotemporal dementia.
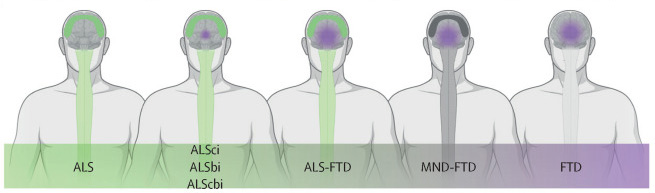
The Lancet, Figure 1 C: amyotrophic lateral sclerosis occurs on a continuum with frontotemporal dementia
5. ALS is put into two categories based on family history.
Familial ALS occurs in patients with a family history of the disease (about 15% of cases), ALS can also occur in patients without a family history of the disease, called “sporadic ALS
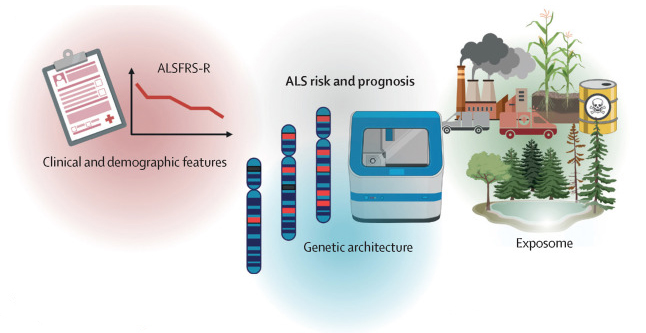
The Lancet, Figure 4 C: a schematic overview of factors that affect amyotrophic lateral sclerosis risk (onset) and prognosis
6. There are over 40 known ALS genes.
The most common mutations are to genes C9orf72, SOD1, TARDBP, and FUS. About 70% of familial and 15% of sporadic ALS cases are associated with a mutation to a known ALS gene.
7. Environmental and occupational hazards are believed to have an impact on ALS risk and progression.
Veterans have a higher-than-normal rate of ALS. Last year, we found that production occupations have a higher risk of ALS due to exposure to toxins.
8. What exactly happens in the body that causes ALS is not completely understood.
However, research suggests that ALS impairs multiple cellular processes, including RNA, protein, and mitochondrial metabolism, trafficking (movement of cargo within neurons), DNA repair, and inflammation.
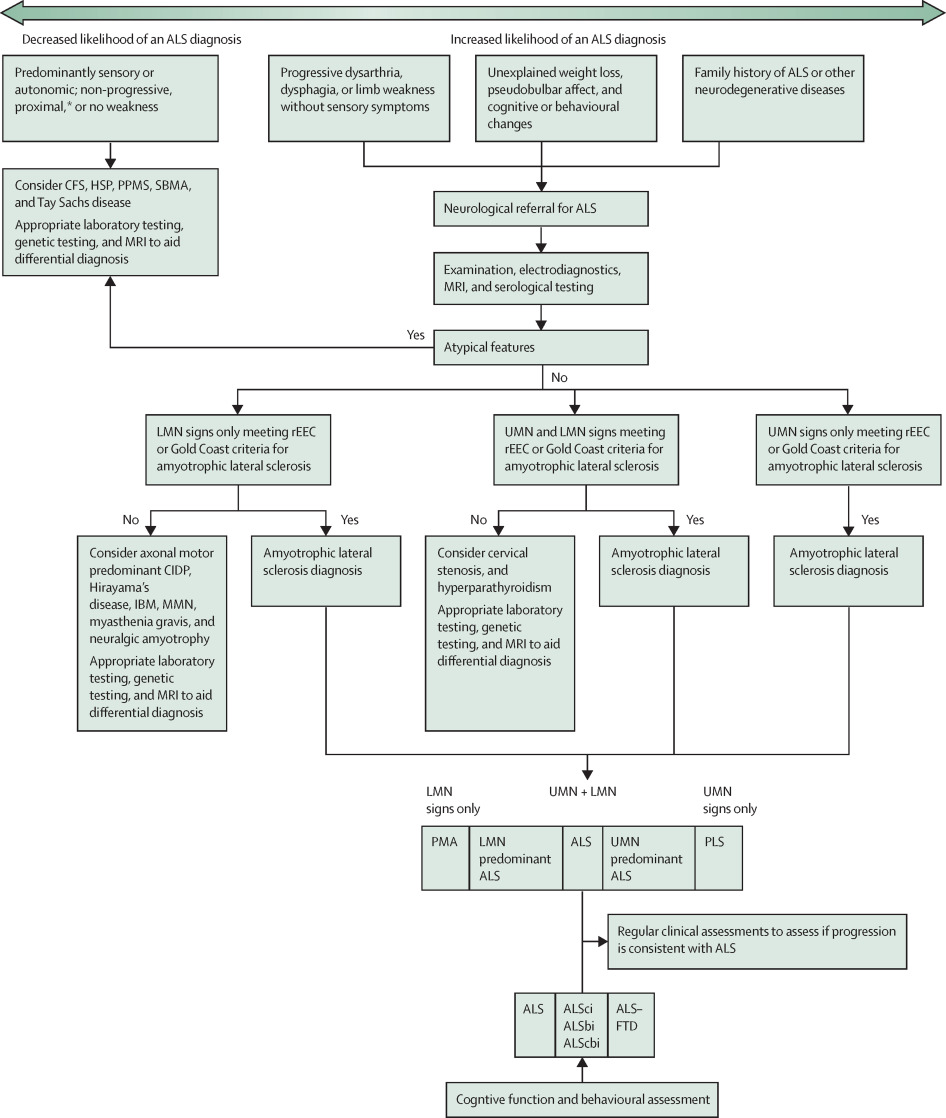
The Lancet, Figure 3: An ALS differential diagnosis chart
9. ALS is incurable.
Treatment is focused on managing symptoms and strategies to improve quality of life. Therapies that can potentially lengthen life in some patients include riluzole, edaravone, non-invasive ventilation, and gastrostomy tube (feeding tube). Research is actively seeking and testing novel therapies.
10. We believe that we can one day make ALS a preventable disease.
(not in The Lancet)





